How much does over-digestion affect input normalization in ChIP-seq?
2022 Jun 1
ChIP
Normalization
R
Fragmented genomic DNA is ideally to appear in the gradient of mono / di / tri-nucleosomes, between 100 and 1000 bp, and centers at 200-300 bp. And pulldown enriches the fragments that can carry the protein of interest, e.g. H3K4me3 occupancy, once or multiple times (but specifying the exact number is complicated). Regarding digestion technical variation, there are two extreme situations, either the uneven fragmentation meets with sparse occupancy, or the genomic DNA is over-fragmented. But how the unbalanced fragmentation affects input normalization results, and if it would introduce a bias, are still unclear.
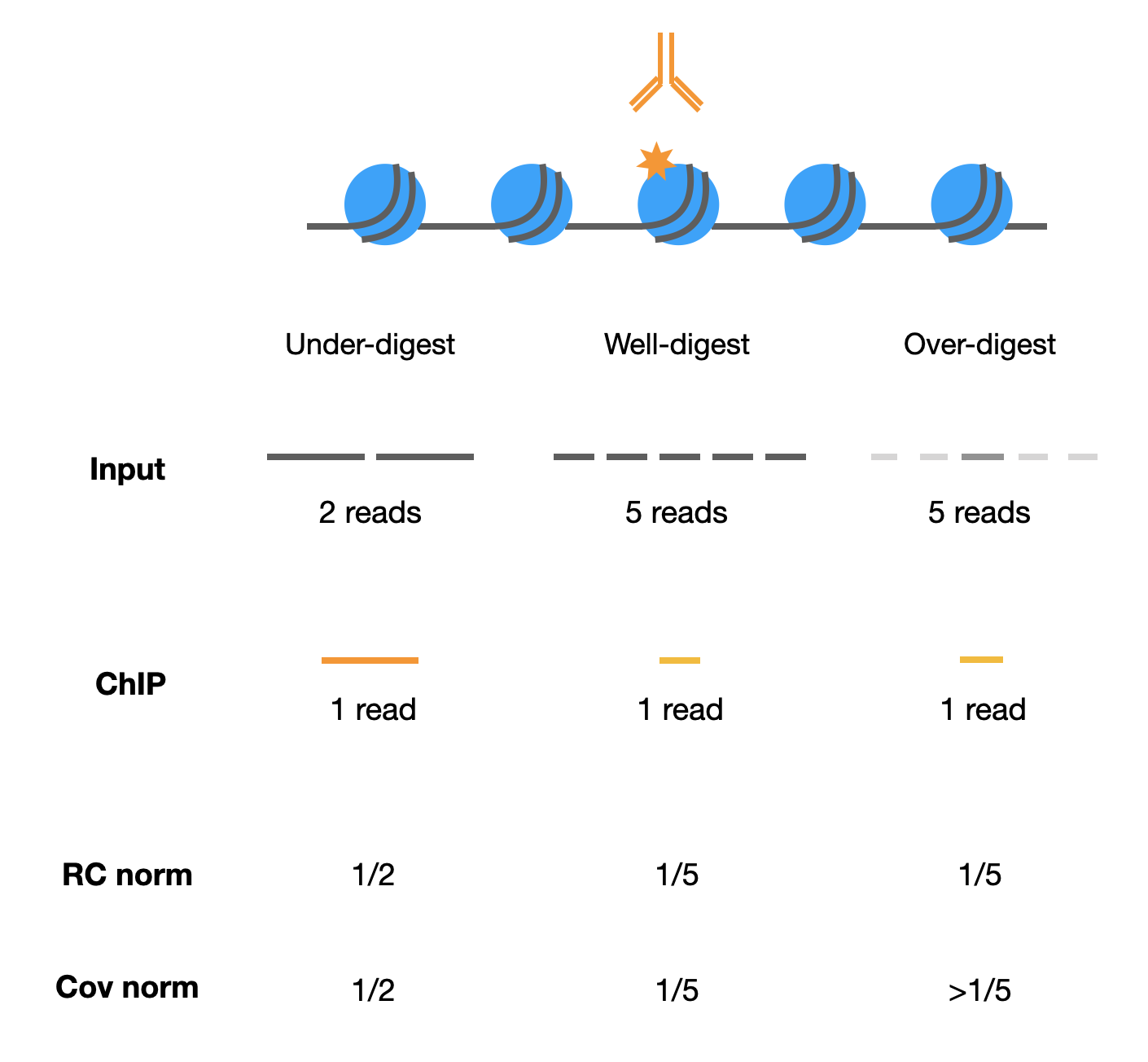
Genomic DNA fragmentation could affect input normalization results. (RC: read count; Cov: coverage)
Given the conceptual cases above, here I will explore the impact of over-digestion on H3K4me4 input normalization. And in the real world, uneven fragmentation is not rare, due to:
- Variation in digestion conditions, e.g. time, temperature, cell viability, lysis concentration, fix/native chromatin.
- Variation in MNase / sonication sensitivity in different genomic regions.
Over-digestion decreases fragment size around the nucleosome, which is the minimal unit of genomic DNA. Compared to the well-digestion, the read counts from the same input amount will be the same.
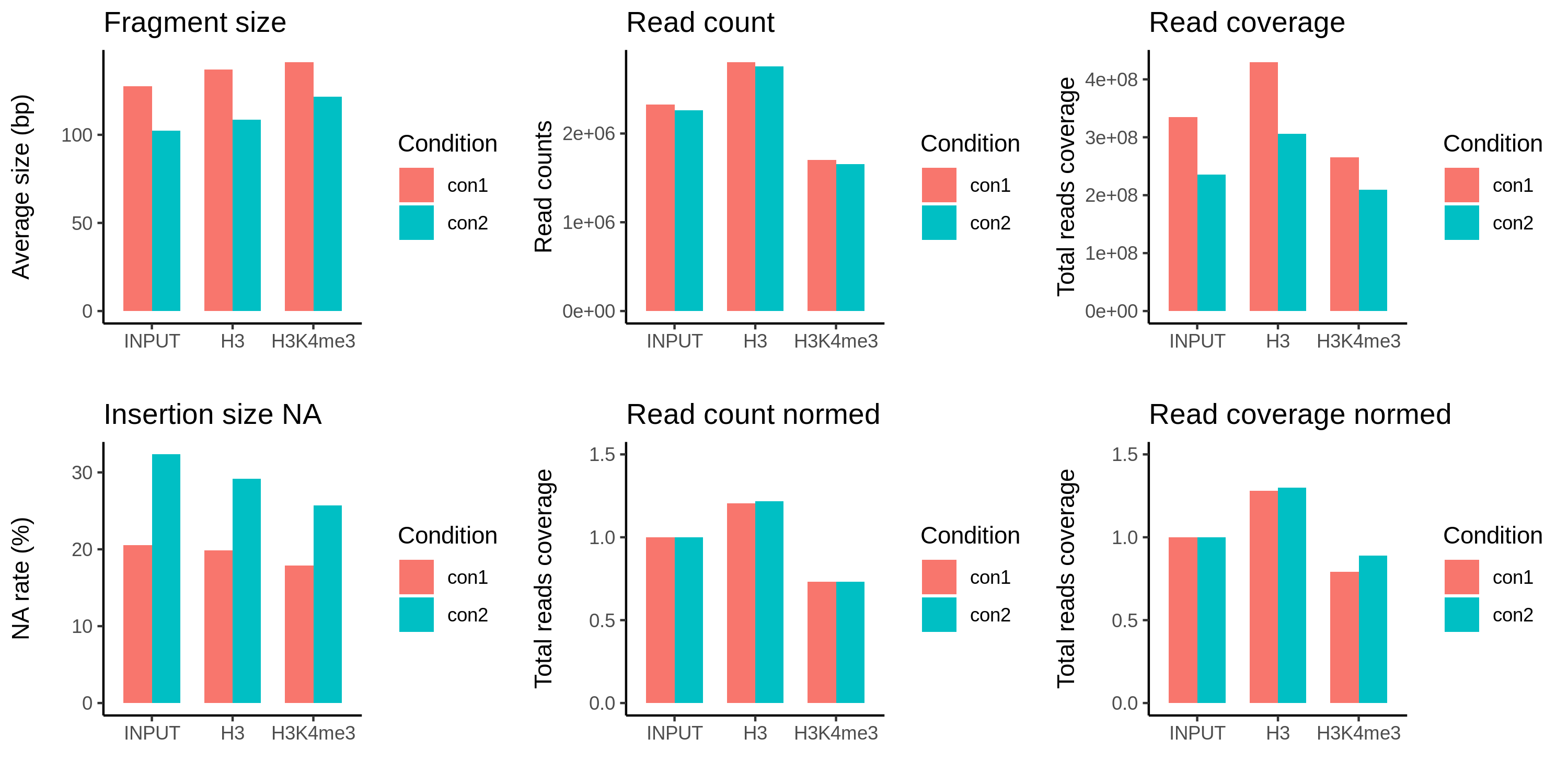
Overview of two conditions of fragmentation, condition1 is well-digested, condition2 is over-digested.
As shown above, over-digestion causes many sub-nucleosomal fragments and unpaired reads, although the total paired reads are unchanged between the conditions. In this case, coverage normalization is no longer suitable in contrast to the under-digested situation.
In addition to the digestion variation between conditions, sample internal bias also exists due to the uneven sensitivity to MNase. Early DNA replication regions are enriched with H3K4me3, and the late regions are the opposite. And the early regions yield larger fragments after MNase digestion.
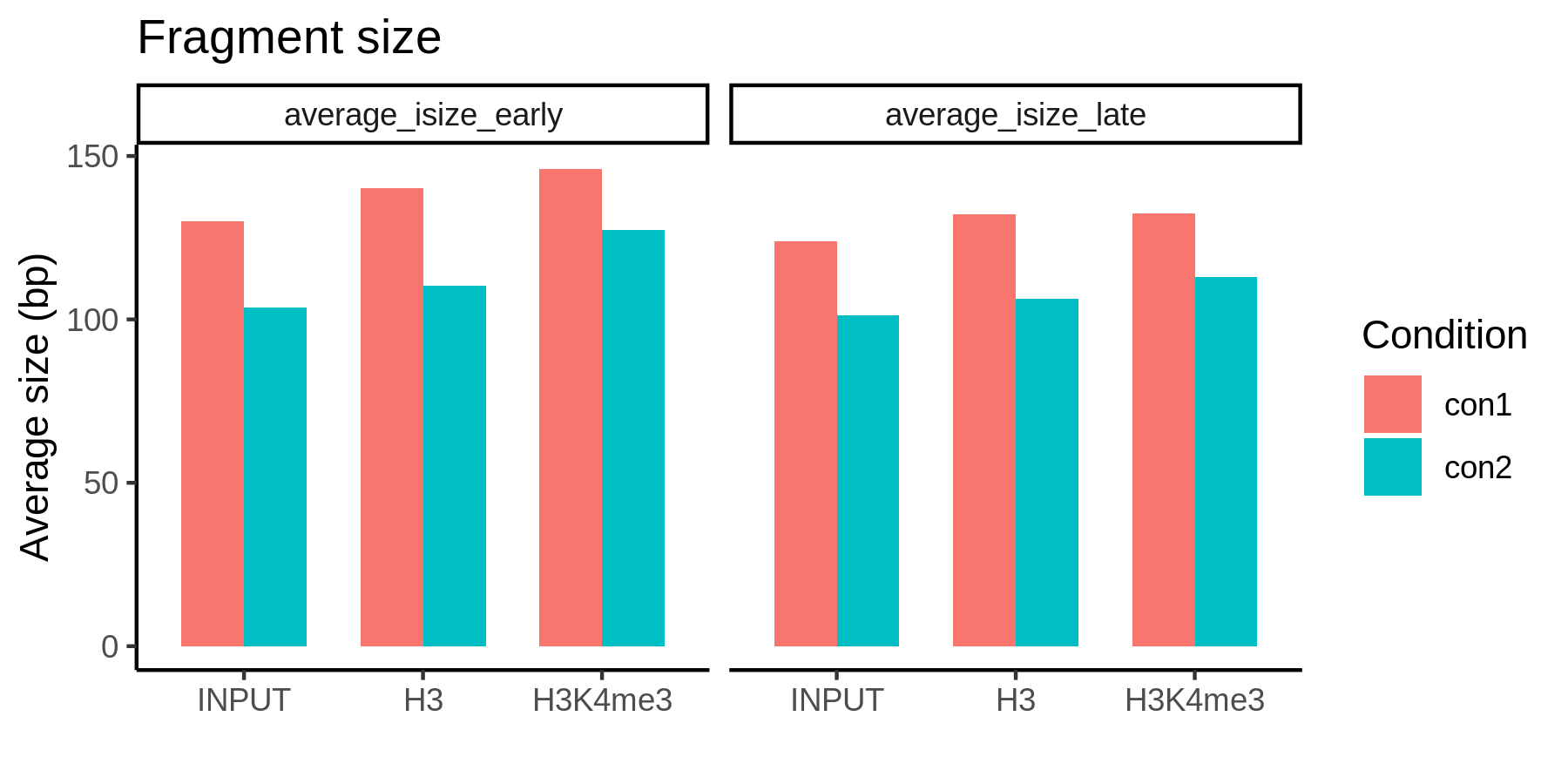
Early and late DNA replication regions have different MNase sensitivity.
Since H3K4me3 bound fragments are larger than the average input fragment size, coverage normalization will introduce a technical bias that is specific to the H3K4me3 targets (early regions). For the well-digested condition, coverage normalization will enlarge the dispersion between enriched / depleted regions.
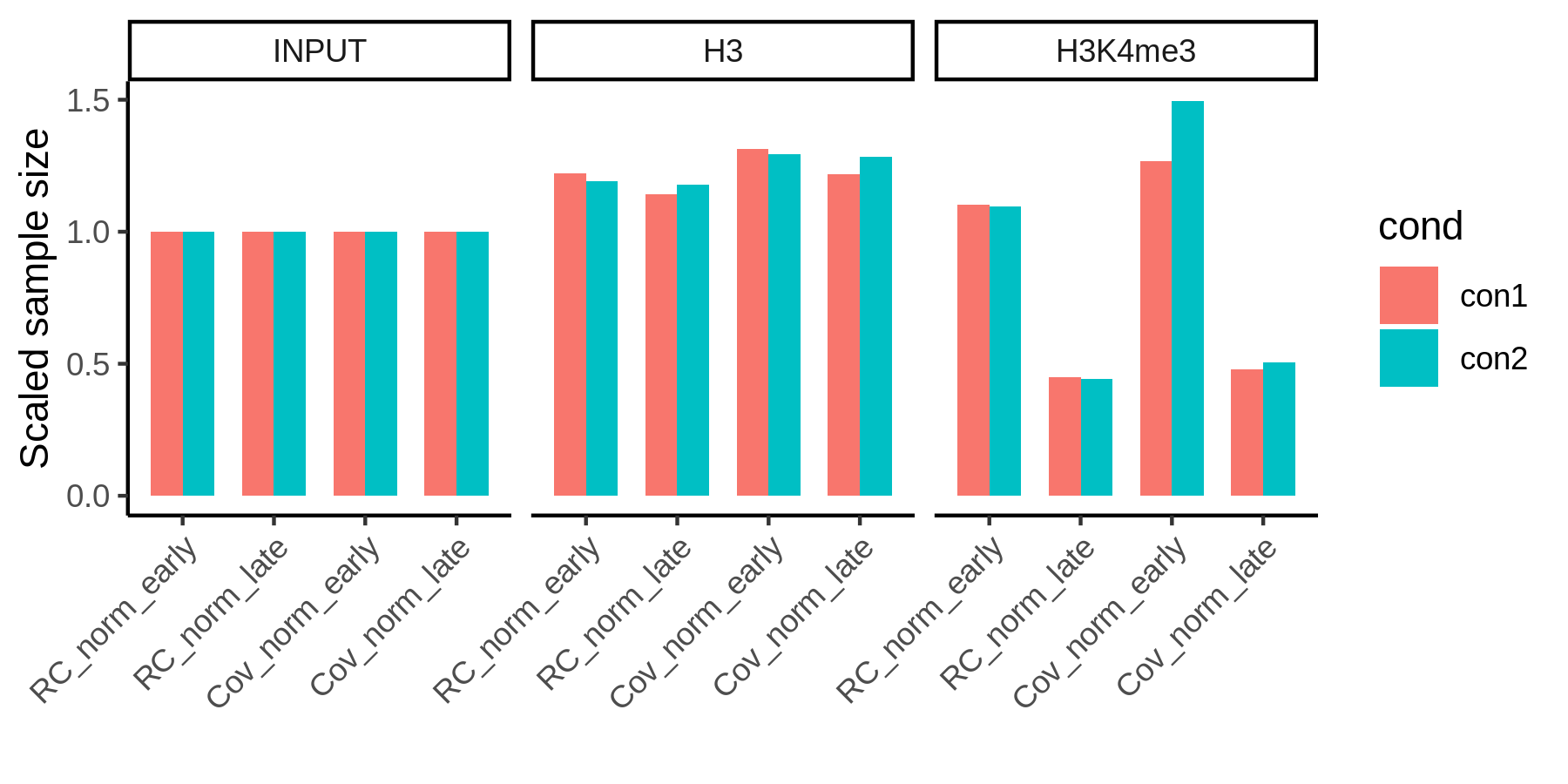
Coverage normalization has a bias for the target enriched regions.
Therefore, over-digestion has a minimal impact on sample size with the read count normalization.